But first, some background info. PKU, otherwise known as Phenylketonuria, that if left untreated, can cause mental retardation. PKU is caused my a mutation on chromosome 12. To pass PKU on to later generations, both parents must have one out of their two PAH (Phenylalanine hydroxylase, an enzyme that catalyses the reaction responsible for the addition of a hydroxyl group to the end of the 6-carbon aromatic ring of phenylalanine, such that it becomes tyrosine)
 |
| Phenylalanie |
 |
| Tyrosine |
Because PKU occurs in 1 in 10,000 to 15,000 newborns, most babies in the US are screened for PKU shortly after birth. This is done by pricking the heel or hand of the baby and collecting the drops on a Guthrie Card. The blood spot is placed on a plate with a bacteria that cannot grow without PKU. If no bacteria grows then the child does not have PKU. Simple right? If the test is positive further tests are conducted to find if the level of PKU is connected to PAH or the rare (only 2%) defect in the co-factor (BH4) of PAH. he signs and symptoms of PKU vary from mild to severe. The most severe form of this disorder is known as classic PKU. Infants with classic PKU appear normal until they are a few months old. Without treatment with a special low-phenylalanine diet, these children develop permanent intellectual disability. Seizures, delayed development, behavioral problems, and psychiatric disorders are also common. Untreated individuals may have a musty or mouse-like odor as a side effect of excess phenylalanine in the body. Children with classic PKU tend to have lighter skin and hair than unaffected family members and are also likely to have skin disorders such as eczema. As PAH normally converts phenylalanine to tyrosine, the block occurs between phenylalanine and tyrosine. As phenylalanine cannot be converted to tyrosine it builds up in the blood, when it reaches high levels it is termed hyperphenylalaninemia. This disrupted pathway interrupts an entire process, shown below.
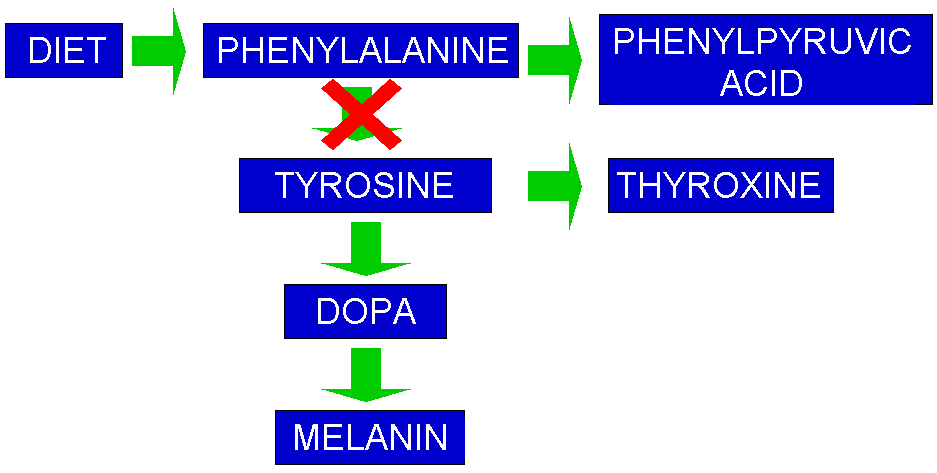
This has been a 30 minute blogpost. All about PKU. Enjoy.
1. What enzyme is most commonly defective in people with phenylketonuria?
2. What reaction does this enzyme catalyze? (What is the substrate and what product is produced?)
3. Describe the symptoms of phenylketonuria.
4. What causes the symptoms of PKU, the lack of a substance or the buildup of one?
5. How common is phenylketonuria? How is it treated?
No comments:
Post a Comment